Ed's Big Plans
Ed's Big PlansArchive for the ‘Andre Masella’ tag
C & Bioinformatics: ASCII Nucleotide Comparison Circuit
Here’s a function I developed for Andre about a week ago. The C function takes two arguments. Both arguments are C characters. The first argument corresponds to a degenerate nucleotide as in the below table. The second argument corresponds to a non-degenerate nucleotide {‘A’, ‘C’, ‘G’, ‘T’} or any nucleotide ‘N’. The function returns zero if the logical intersection between the two arguments is zero. The function returns a one-hot binary encoding for the logical intersection if it exists so that {‘A’ = 1, ‘C’ = 2, ‘G’ = 4, ‘T’ = 8} and {‘N’ = 15}. All of this is done treating the lower 5-bits of the ASCII encoding for each character as wires of a circuit.
| Character | ASCII (Low 5-Bits) |
Represents | One Hot | Equals |
| A |
00001 |
A | 0001 |
1 |
| B | 00010 | CGT | 1110 |
14 |
| C | 00011 | C | 0010 |
2 |
| D | 00100 | AGT | 1101 |
13 |
| G | 00111 | G | 0100 |
4 |
| H | 01000 | ACT | 1011 |
11 |
| K |
01011 | GT | 1100 |
12 |
| M | 01101 | AC | 0011 |
3 |
| N | 01110 | ACGT | 1111 |
15 |
| R | 10010 | AG | 0101 | 5 |
| S | 10011 | GC | 0110 |
6 |
| T | 10100 | T |
1000 |
8 |
| V | 10110 | ACG |
0111 |
7 |
| W | 10111 | AT |
1001 |
9 |
| Y | 11001 | CT |
1010 |
10 |
The premise is that removing all of the logical branching and using only binary operators would make things a bit faster — I’m actually not sure if the following solution is faster because there are twelve variables local to the function scope — we can be assured that at least half of these variables will be stored outside of cache and will have to live in memory. We’d get a moderate speed boost if at all.
/*
f():
Bitwise comparison circuit that treats nucleotide and degenerate
nucleotide ascii characters as mathematical sets.
The operation performed is set i intersect j.
arg char i:
Primer -- accepts ascii |ABCDG HKMNR STVWY| = 15.
arg char j:
Sequence -- accepts ascii |ACGTN| = 5.
return char (k = 0):
false -- i intersect j is empty.
return char (k > 0):
1 -- the intersection is 'A'
2 -- the intersection is 'C'
4 -- the intersection is 'G'
8 -- the intersection is 'T'
15 -- the intersection is 'N'
return char (undefined value):
? -- if any other characters are placed in i or j.
*/
char f(char i, char j) {
// break apart Primer into bits ...
char p = (i >> 4) &1;
char q = (i >> 3) &1;
char r = (i >> 2) &1;
char s = (i >> 1) &1;
char t = i &1;
// break apart Sequence into bits ...
char a = (j >> 4) &1;
char b = (j >> 3) &1;
char c = (j >> 2) &1;
char d = (j >> 1) &1;
char e = j &1;
return
( // == PRIMER CIRCUIT ==
( // -- A --
((p|q|r|s )^1) & t |
((p|q| s|t)^1) & r |
((p| r|s|t)^1) & q |
((p| s )^1) & q&r& t |
((p| t)^1) & q&r&s |
(( q| t)^1) & p&s |
(( q )^1) & p&r&s
)
|
( // -- C --
((p|q|r )^1) & s |
((p| r|s|t)^1) & q |
((p| s )^1) & q&r& t |
((p| t)^1) & q&r&s |
(( q|r )^1) & s&t |
(( q| t)^1) & p &r&s |
(( r|s )^1) & p&q& t
) << 1
|
( // -- G --
(( q|r| t)^1) & s |
((p|q| s|t)^1) & r |
((p|q )^1) & r&s&t |
((p| r )^1) & q& s&t |
((p| t)^1) & q&r&s |
(( q|r )^1) & p& s |
(( q| t)^1) & p& s
) << 2
|
( // -- T --
((p|q|r| t)^1) & s |
(( q| s|t)^1) & r |
((p| r|s|t)^1) & q |
((p| r )^1) & q& s&t |
((p| t)^1) & q&r&s |
(( q )^1) & p& r&s&t |
(( r|s )^1) & p&q& t
) << 3
)
&
( // == SEQUENCE CIRCUIT ==
( // -- A --
((a|b|c|d )^1) & e |
((a| e)^1) & b&c&d
)
|
( // -- C --
((a|b|c )^1) & d&e |
((a| e)^1) & b&c&d
) << 1
|
( // -- G --
((a|b )^1) & c&d&e |
((a| e)^1) & b&c&d
) << 2
|
( // -- T --
((a| e)^1) & b&c&d |
(( b| d|e)^1) & a& c
) << 3
);
}
Andre’s eventual solution was to use a look-up table which very likely proves faster in practice. At the very least, this was a nice refresher and practical example for circuit logic using four sets of minterms (one for each one-hot output wire).
Should you need this logic to build a fast physical circuit or have some magical architecture with a dozen registers (accessible to the compiler), be my guest 😀
BioCompiler might start life as BioCOBOL
Update: Matthew has found an even more thorough review paper that discusses computer assisted synthetic biology approaches — it can be found at doi:10.1016/j.copbio.2009.08.007.
The iGEM competition year is running to a close. The teams are headed into November 2010 and have roughly one month left before attending the conference. I’m personally not attending the conference this year — I think the undergraduates will get more out of the experience.
The current year sees our continued efforts to precipitate a dedicated software team — a functioning autonomous unit that will serve to supplement and enhance Waterloo’s impact in the iGEM competition. More importantly, we’re going to do some very interesting science. We’ve had some success talking with other student groups across campus — notably, we should probably talk with the student IEEE/CUBE chapter when we have more work completed. We had involved BIC as well, however, it was early on and we had even less ground to stand upon ( — the primordial software team was mostly interested in BioMortar and BrickLayer at the time — the later project having been taken up by the iGEM Coop students as in Python this year).
This whole BioCompiler business started when Matthew uncovered a nice candidate problem: the compilation of a schematic for behaviour to a fully functioning synthetic biological circuit. Let’s be as precise as we can be here. I mean to say, we will take a description (which could resemble a piece of formal language source code) — and have it compiled into the sequence of BioBricks that will produce the desired behaviour.
This idea has been approached by several groups before — but each time, a different subproblem was considered (this is a reorganization of Matthew’s very nice list here).
- Synthetic biology programming language: Genetic Engineering of Living Cells (GEC) (Microsoft Research) is a project that hones in on a formal language specification.
- Synthetic biology computer assisted design (CAD): Berkeley Software (iGEM 2009) created a suite of items — Eugene (formal language for synthetic biology), Spectacles (visualizer integrating parts with their behaviours), Kepler (a dataflow broker). As well, Berkeley is responsible for the award winning Clotho (iGEM 2008 – Best Software Tool) which is a workbench that connects with the parts registry database (amongst other possible resources).
- Systems biology pathway reaction simulation: Systems Biology Markup Language (SBML), Systems Biology Workbench (SBW) and Jarnac are a set of tools that perform systems biology analysis (which we consider to be an output of synthetic biology). SBML is the formal language, Jarnac is a reaction network simulator (which utilizes JDesigner as a front end) and SBML is a dataflow broker between SBML and Jarnac.
- BioBrick specific pathway simulation: Minnesota’s Team Comparator (iGEM 2008) created SynBioSS — a tool which estimates the concentration of reactants and products given the appropriate BioBricks on a simulated circuit.
Update — here are a few more items thanks to Matthew Gingerich, George Zarubin and Andre Masella.
- Molecular biology and bioinformatics analysis: The European Molecular Biology Open Software Suite (EMBOSS) is a toolkit developed by the European Molecular Biology Network (EMBnet) for bioinformatics. This might not be immediately relevant, but it is interesting. EMBOSS is actually relatively complete. More distantly along this vein, there’s also Bioconductor which focuses mostly on microarray analysis and is implemented in R.
- More synthetic biology computer aided design (CAD): TinkerCell is a GUI-driven piece of software that supports extension with C++ and Python. TinkerCell along with the suite created by Berkeley Software are the two most promising target systems in which to integrate BioCompiler. Finally, there’s GenoCAD which appears to be in an early phase of development — this software looks to emphasize construction with correct syntax and attribute grammars — I’ll have to read more about this.
- Formal laboratory protocol description: BioCoder is another Microsoft research project — the designed language aims to be both human readable and complete for automation. It reminds me of standard operating procedures (SOP) with greater precision. While BioCoder compiles from protocol to automation, we’ll be compiling to circuitry and protocol. BioCoder will give us some insights about the kinds of protocols that others are thinking of.
There is of course more software, but these are the items that we have become most familiar with — that we like — and that we consider to be standards toward which our own work should strive.
Two interesting problems arise when we think about these subproblems. First, the programming languages specified (including Eugene from Berkeley’s CAD suite) are exactly what they claim to be. Formal specifications. This is possible because of how concrete they are. They literally document what a synthetic biology circuit is. But this isn’t too different from what humans have been doing in iGEM all along. Second, the synthetic biology items don’t really seem to talk to the systems biology items — whereas we expect that the two — being input and output — to be inextricably linked. I explain my thoughts on the two below.
Why a programming language?
Andre enlightened me to this the other day. Humans invented programming languages to do two things. On an arrow running from the concrete toward the esoteric, we have the practical concern of compressing the amount of code that we want to write while retaining our programs’ expressiveness. This is the origin of macro systems such as COBOL. On an arrow pointing in the reverse — from the cerebral abstract down to the literal — we have the theoretical concern of mathematical beauty, of completeness. This is the origin of such functional languages like Lisp. For humans to have any hope of creating such a Lisp-like language for synthetic biology — we would need to understand all of the reactionary nuances about it, the system with which we tamper — at least inasmuch as a painful heuristic approximation. This is a feat we are no where near completing though footholds are managed with systems biology.
So here we are, BioCOBOL is the first step. A developing simple — though complete macro-like system that is in league in terms of abstraction with the programming language / CAD -like projects we’ve seen thus far. Only, we aim to increase the efficiency of circuit design; so that the programming language is not a literal mapping of the human document — rather, it is an explanation of behaviour. We will abstract it ever so slightly with each iteration of development — departing further and further away from CAD. A subteam headed by Brandon is currently developing the syntax and search algorithms required for the job — Brandon suggests that a weighted traversal of valid circuits should form our algorithmic primitive. My subteam is attempting to characterize as many known circuits in iGEM as possible — analyzing what pieces of input (stimuli: chemical concentrations, gradients, quora, oscillators) may be compressed for their common usage — what function prototypes already exist ( — reacts to an input: promoters) for compression — what the standard outputs are (analogy to printing error messages: GFP-family) etc. — again in the effort to realize what is losslessly compressible. Our software should eventually provide the correct laboratory protocol as well.
In this sense, we are respecting what a compiler is before we even approach more sophisticated compilation: it transforms a document in one language to another.
Incidentally, I should mention that Jordan and George are working on a modelling problem with the lab team — I’m not clear on the specifics, but I take it they want to pull out some differential equations on a set of promoters.
Why do we care that Synthetic Biology logic should talk to Systems Biology logic?
Finally, it is clear that we will eventually want to become even more abstract — even more mathematically complete, even more expressive. While we may never know enough about systems biology to create BioLISP (in our lifetime), we expect there to be sufficient research for us to discover — and perhaps research we can conduct ourselves to come ever closer. Systems biology allows us to think about synthetic biology in terms of reaction concentrations; free energy etc.. It gives the notion of compilation its own ground; the ground we want to cover. Imagine the perfect BioCompiler — stating the a problem to be compiled in terms of the input and output of the system. Let’s be precise here: I mean to say, the products and reactants or behaviour of our circuit. Let us describe what our circuit will do instead of what our circuit is made of. This — the missing link, this compiler — is the logical final step of BioCompiler.
Parsing a Newick Tree with recursive descent
>>> Attached: ( parse_newick.js – implementation in JS | index.html – example use ) <<<
Update: Fixed parse_newick.js source code that caused a field to be mislabelled in index.html (2010 Dec. 19)
This method of parsing works for any language that allows recursive functions — basically any of the curly bracket languages (C#, C, Java) and the fast-development languages (Ruby, JavaScript, Python) can do this. This particular example will include JavaScript code which you can take and do whatever you want with — recursive descent parsing is a well-known and solved problem, so it’s no skin off my back. Before we begin, I should probably explain what a tree in Newick format is.
What exactly is the Newick format?
Remember back in second year CS when your data structures / abstract data types / algorithms instructor said you can represent any tree as a traversal? Well, if not — don’t fret, it’s not hard to get the hang of.
A traversal is a finite, deterministic representation of a tree which can be thought of as a road map of how to visit each node of the tree exactly once. The phylogeny / bioinformatics people will know each of these nodes as either taxa / sequences (the leaves) or profiles (the internal nodes — i.e. not the leaves). A tree in Newick format is exactly that — a traversal. More specifically, it is a depth-first post-order traversal. When traversing a tree, we have a few options about how we want to visit the nodes — depth-first post-order means we want to start at the left-most, deepest leaf, and recursively visit all of the nodes before we end at the root.
A tree expressed in Newick format describes a tree as an observer has seen it having visited each node in a depth-first post-order traversal.
A node in the Newick format can be…
- A leaf in the form: “<name>:<branch_length>”
- e.g. “lcl|86165:-0“
- e.g. “gi|15072471:0.00759886“
- e.g. “gi|221105210:0.00759886“
- Or a node in the form “(<node>,<node>):<branch_length>”
- This is where it gets tricky and recursive — the “node” item above can be another node, or a leaf — when a node is composed of at least another node, we call that a recursive case; when a node is composed of only leaves, we call that a base case. We make this clearer in the next section.
- Or a node in the form “(<node>,<node>);”
- The semi-colon at the end means that we have finished describing the tree. There is no branch length as the root of the tree is not connected to a parent.
Understanding recursion in a Newick Tree
Base case e.g.: The below node is composed of two leaves.
“(lcl|86165:-0,gi|87118305:-0):0.321158”
Recursive case e.g.: The below node is composed of a leaf on the left, and another node on the right; this right-node is composed of two leaves.
“(gi|71388322:0.0345038,(gi|221107809:0.00952396,gi|221101741:0.00952396):0.0249798):0.0111081“
If the names look a bit funny, that’s because these are sequences pulled out of a blastp search.
The Newick tree itself may have any number of nodes, the base case being a single node. The only restriction is that any node either is a leaf (has zero children) or must have two children (never a single child). These two children again are allowed to be either leaves or other nodes.
We would visualize the above Recursive case e.g. as follows.:

Where the numbers beside the edges are edge length and the names of the named taxa are indicated inside the circles (leaves). The unnamed internal nodes are the profiles that have been calculated with alignments. You’ll notice that there’s a branch length leading out of the root of this subtree into the rest of the tree (not shown). If this was a complete tree on its own, the root would have no branch length there and no arrow coming out of it.
Finally, Parsing!
Now that we have reviewed the depth-first post-order traversal, the Newick format and how to interpret it; we can move onto parsing these things. In parsing, we hope to create an in-memory structure that represents the tree. The parser I discuss assumes that the Newick format string already has had all newlines stripped, as it uses regular expressions. If your trees aren’t stored that way, just make sure that newlines are stripped before feeding it into my function. The in-memory object that’s created will have the following fields as a minimum.
- node_type $left – the left child of a node
- node_type $right – the right child of a node
- string_type $name – the name of a node (if it is a leaf)
- float_type $branch_length – the length of the branch coming out of the top of a given node
I also use the following two fields to keep track of a few house keeping items and to make life pleasant for future tasks with this in-memory structure.
- node_type $parent – the parent of a node
- integer_type $serial – the order in which this node occurs when parsed
Note that in future, a depth-first post-order traversal will yield the nodes in the exact order that they were read — so in reality “this.serial” isn’t really needed, but it’s nice to have for future functions you might write as a shorthand. Have you understood why these nodes would occur in the same order?
Each language has its own quirks. Python and JavaScript have the advantage that you can define a class and define its properties (fields) on the fly — meaning there’s less code to download and to write, which is particularly good for blogs 😛
Here’s pseudo-code for the recursive descent parsing function “tree_builder(<newick_string>)” — I don’t indicate where to strip away newlines because I assume they’re gone. I also don’t indicate where to strip away whitespace (I did previously, but that took up like half the pseudocode). So if there’s a place where you suspect whitespace can occur, strip it away and increment the cursor by the length of that whitespace.
Note: We are assuming that there are no newlines in the file — that is, the entire newick traversal is one string on a single long unbroken line.
// Note:
// Call tree_builder() with a "new" operator in JavaScript
// to allow it to use the 'this' keyword.
// In other languages,
// you may need to explicitly create a new object instead of using 'this'.
var count = 0; // can be either a global variable or a reference
// keeps track of how many nodes we've seen
var cursor = 0; // also either a global or a reference
// keeps track of where in the string we are
function tree_builder(newick_string) {
// Look for things relating to the left child of this node //
if $newick_string [ $cursor ] == "(" {
$cursor ++; // move cursor to position after "(".
if newick_string [ $cursor ] == "(" {
// Then the $left node is an internal node: requires recursion //
$this.left = new tree_builder(newick_string); // RECURSIVE CALL.
$this.left.parent = $this;
}
// try to find a valid $name next at the current cursor position //
if $name = $newick_string [ $cursor ] . regex match ("^[0-9A-Za-z_|]+") {
// Then the $left node is a leaf node: parse the leaf data //
$this.left = new Object;
$this.left.name = $name;
$this.left.serial = $count;
$count ++;
$this.left.parent = $this;
$cursor += length of $name;
// move cursor to position after matched name.
}
// notice: if no $name found, just skip to finding branch length.
}
if $newick_string [ $cursor ] == ":" {
// Expect left branch length after descending into the left child //
$cursor ++; // move cursor to position after the colon.
// look for the string representation of a floating point value "FPV"...
$branch_length_string =
$newick_string [ $cursor ] . regex match ("^[0-9.+eE-]+");
$this.$left.$score = $branch_length_string as a FPV;
$cursor += length of the string $branch_length_string;
// move cursor after FPV.
}
// Look for things related to the right node //
if $newick_string [ $cursor ] == "," {
$cursor ++; // move cursor after the comma
if newick_string [ $cursor ] == "(" {
// Then the $right node is an internal node: requires recursion //
$this.right = new tree_builder($newick_string); // RECURSIVE CALL.
$this.right.parent = $this;
}
// try to find a valid $name next at the current cursor position //
if $name = $newick_string [ $cursor ] . regex match ("^[0-9A-Za-z_|]+") {
// Then the $right node is a leaf node: parse the leaf data //
$this.right = new Object;
$this.right.name = $name;
$this.right.serial = $count;
$serial ++;
$this.right.parent = $this;
$cursor += $length of $name;
}
// again, accept if no $name found and move onto branch length.
}
// Now looking for the branch length ... //
if $newick_string at $cursor == ":" {
// Expect right branch length after descending into right child //
$cursor ++; // moving past the colon.
$branch_length_string =
$newick_string [ $cursor ] . regex match ("^[0-9.+eE-]+");
$this.right.score = $branch_length_string as a FPV;
$cursor += length of the string $branch_length_string;
}
if $newick_string at $cursor == ")" {
$cursor ++; // move past ")".
}
// Expect the branch length of $this node after analyzing both children //
if $newick_string at $cursor == ":" {
$cursor ++;
$branch_length_string =
$newick_string [ $cursor ] . regex match ("^[0-9.+eE-]+");
$this.score = $branch_length_string as a FPV;
$cursor += length of the string $branch_length_string;
}
if $newick_string at $cursor == ";" {
// This case only executes for the root node-- //
// --the very last node to finish parsing //
$cursor ++; // to account for the semicolon (optional, consistency)
$this.serial = $count; // no need to increment $count here
}
// Return to complete a recursive instance (internal) or base case (leaf) //
return this; // not needed if "new" operator was used all along
}
The implementation that’s included in the attached JavaScript does two things differently than the above. First, I don’t force the user to break encapsulation by declaring globals. Instead, I use an object called “ref” which has two fields, “ref.count” and “ref.cursor”. The name “ref” is chosen to remind us that its fields are passed by reference. This is possible because the object “ref” itself is passed by reference (I could have called it anything, but I chose a name that suited its purpose). Imagine trying to pass “count” and “cursor” in this recursive function without an enclosing object — what would happen instead is that the value wouldn’t be updated, instead the integer values would be copied and modified locally in the span of single function instances. The “ref” object is passed as an argument to the “_tree_builder(<ref>, <newick_string>)” function internally where it is instantiated if “null” is passed in the place of “ref”. The calling user doesn’t even need to know about it, as it’s all wrapped together in a pretty package and the user just calls “tree_builder(<newick_string>)” as before — without the need for globals. The enclosing function even does the work of instantiating the root node with the “new” keyword so that the end user doesn’t even need to (and shouldn’t!) say “new”.
Finally, there is a traversal function, and an expose function. The traversal function returns the node objects in an array following in the natural depth-first post-order described previously. The expose function is a debugging function which writes the output using “thoughts_write(<message>)” which wraps “document.write(<message>)”. You can change “thoughts_write(<message>)” to print output elsewhere using “document.getElementById(<some_identity>).innerHTML += (<message>)” where “some_identity” is the “id” of some element in your HTML. Without going into detail, the traversal function also uses an internal “ref” object which refers to the array as it is filled.
Some other things you can try…
The JavaScript recursive descent parser is actually a simplified version of something that I threw together earlier. The major difference is that I tag leaf nodes with fasta protein sequences by passing a reference to an associative array (or hash) as an argument to “tree_builder(<newick_string>, <fasta_hash>)”. Remember, in JavaScript, an associative array is an instance of an Object, not an instance of an Array. Using non-integer field names in an Array is actually not well defined but kind of sort of works semi-consistently between browsers (i.e. don’t try!), but that’s a story for another day. Whenever the name of a leaf is parsed, I would look it up in the hash and put it in a field either “this.left.fasta” or “this.right.fasta” thereby conferring to the leaf nodes of the in-memory structure the appropriate fasta protein sequence.
Lessons Learned
In putting this thing together, I managed to relearn JS, recursive descent parsing and making JS pass things by reference up and down a recursive stack of function calls. Andre Masella helpfully reminded me that I didn’t need to use any kind of BNF or LALR(1) parser — just an easy single-function parser would do. This is actually a port of a C# parser which does the same thing that I wrote in an earlier step for phylogenetic analysis — the difference being that C# offers a “ref” keyword that allows one to specify arguments in a function’s parameter list as emulated references (functionally equivalent to using C’s asterisk and ampersand convention though not operationally equivalent).
Happy parsing! The code presented should be easy enough to understand for you to port it into the language of your choice for whatever purposes you design.
Apache Optimized Finally! (Firebug, YSlow)
I didn’t realize I hadn’t added the mod_expires.c and mod_deflate.c items to my httpd.conf file in Apache yet– Andre clued me in!
Andre noticed my blog was taking a while to load, even when the browser cache should have significantly dented the page weight. He used Firebug and Yahoo’s YSlow to make a diagnosis and told me to do the same– this page ended up taking a whopping 17 seconds to load which is … very … sad. After I added these lines to my httpd.conf file, things were looking better (roughly 1.5 seconds — not perfect, but it’s far better).
The mod_expires.c chunk specifies that files displayed on a webpage ought to live in the browser cache. The caching information is sent as part of the file header by Apache to the client browser. Without this, files were apparently expiring instantly meaning that each refresh required downloading every single file again including including the images comprising this theme’s background.
The mod_deflate.c chunk specifies that file data should be gzipped before transmitting– this is again handled by Apache. The trade off between compressing a few text files (even dynamically generated ones) versus sending uncompressed text is more than fair.
<IfModule mod_expires.c>
FileETag MTime Size
ExpiresActive On
ExpiresByType image/gif "access plus 2 months"
ExpiresByType image/png "access plus 2 months"
ExpiresByType image/jpeg "access plus 2 months"
ExpiresByType text/css "access plus 2 months"
ExpiresByType application/js "access plus 2 months"
ExpiresByType application/javascript "access plus 2 months"
ExpiresByType application/x-javascript "access plus 2 months"
</IfModule>
<IfModule mod_deflate.c>
# these are known to be safe with MSIE 6
AddOutputFilterByType DEFLATE text/html text/plain text/xml
# everything else may cause problems with MSIE 6
AddOutputFilterByType DEFLATE text/css
AddOutputFilterByType DEFLATE application/x-javascript
AddOutputFilterByType DEFLATE application/javascript
AddOutputFilterByType DEFLATE application/ecmascript
AddOutputFilterByType DEFLATE application/rss+xml
</IfModule>
I’ve also removed the custom truetype font files specified in the CSS… they aren’t handled correctly for whatever reason– even after I added ‘font/ttf’ entries to the mod_expires.c chunk above. Finally, I tried completely removing background images from the site and restoring them again– it doesn’t make things any faster after images have been cached (correctly, finally).
I am very happy.
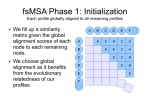
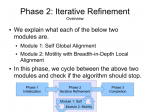
fsMSA Algorithm… Monkeys in a β-Barrel…
I’ve finally finished documenting my foil sensitive protein sequence algorithm… This is part of Monkeys in a β-Barrel — a work in progress, this time continuing more on Andrew’s half of the problem rather than Aron’s.
I’ve decided on using the word “foil” to mean “internal repeat” since it’s easier to say and less awkward in written sentences. Andre suggested it after “trefoil” and “cinquefoil”, the plant.
Thumbnails below (if you are curious about the full slide show, contact me :D).