 Ed's Big Plans
Ed's Big PlansArchive for the ‘Aron Broom’ tag
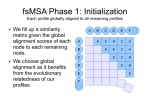
fsMSA Algorithm… Monkeys in a β-Barrel…
I’ve finally finished documenting my foil sensitive protein sequence algorithm… This is part of Monkeys in a β-Barrel — a work in progress, this time continuing more on Andrew’s half of the problem rather than Aron’s.
I’ve decided on using the word “foil” to mean “internal repeat” since it’s easier to say and less awkward in written sentences. Andre suggested it after “trefoil” and “cinquefoil”, the plant.
Thumbnails below (if you are curious about the full slide show, contact me :D).

Protein Project Progress…
Last week, Liz, Aron, Andrew, Brendan and I sat down to discuss the beta-trefoil project. It was a good chance for me to understand the methods used and the kinds of results we are interested in for my own TIM Barrel project.
Continuing on with the structural repeat problem, I’ll today be writing a short FSA parser that can handle DSSP or DSS output– simply, a very primitive machine will be used to imitate a human’s visual inspection of repeated secondary structural elements in given proteins. This is in line with the work I did manually staring at structures to get a grasp of how to look at protein models, and also in line with the objective to automate much of this work. Prior to that step, I reduced the probability of doing redundant work by using BLASTCLUST and selecting only a few known structures in each cluster to inspect… a sequence based alignment for each cluster will inform me of where my manually detected repeat boundaries map to the remaining sequences.
Oddity: If you BLASTCLUST all the “FULL” (not “SEED”) sets of TIM Barrel sequences for the entire fold from PFAM along with the sequences of known TIM Barrel fold structures of SCOP, you’ll find that cluster fifteen (as of today) has these elements:
1YBE_A A6U5X9 A6WV52 Q2KDT0 Q2YNV6 Q6G0X7 Q6G5H6 Q8UIS9 Q8YEP2 Q92S49 Q98D24 A1UUA2
In the above listing, 1YBE:A (PDB code) is the sole known PDB structure, while the remainder are putative TIM Barrels (uniprot codes) as determined by the HMM model from PFAM.
The enzyme 1YBE looks like this…
It’s an oddity because of the number of alpha-helices inserted within what is usually a hydrophobic beta barrel– the red pieces of ribbon should form a hollow cylinder, but it’s split apart for 1YBE and accommodates a bunch of cyan helices. Labeled in white are helices that break with the beta-alpha repeating secondary structural element (SSE) pattern by occurring before the first repeat. Labeled in green are breaks between beta-alpha SSE patterns.
Reference
Seetharaman, J., Swaminathan, S., Crystal Structure of a Nicotinate phosphoribosyltransferase [To be Published]
TIM Barrels and 4-Alpha Helix Bundles
Beta-Alpha TIM Barrels and 4-Alpha Helix bundles are the first of the major folds I’ll be looking at here at Waterloo…
As with all academic projects, the probability of goal, approach and method mutation is high. Looking at the above protein folds serves as an excellent starting point as I’ll be applying some of the established methods that Andrew and Aaron have developed.
Alpha helices and Beta sheets are objects that any highschool biologist is acquainted with. To recapitulate, alpha helices are sequences of amino acids arranged so that the alpha carbon of each amino acid falls along the path of a helix. The number of amino acids per turn in this peptide and the regularity of the helix are determined by both the sequence and the environment that peptide finds itself in. Amino acids in the beta sheet conformation are arranged so that their alpha carbons zig zag. The result is a nice wide and flat shape schematically drawn as a sheet. Alpha helices and Beta sheets are collectively called secondary structural elements.
So, folds are these giant overarching classification of proteins– Folds themselves are inherently structural, so classifying them OR using them as classifications is only relevant in structural studies and databases on the web like SCOP and CATH. In databases such as SCOP and CATH, classification of similarly structured proteins start by determining whether the protein contains mostly alpha helices; mostly beta sheets; beta sheet and alpha helices alternatively and irregularly; or beta sheet and alpha helices in distinct regions of a protein. In SCOP, further classification is done by manually assigning proteins to smaller and smaller categories, while in CATH, these classifications are done by a hidden Markov model and then manually inspected (or not). It turns out that CATH uses a similar manual approach, and uses HMMs only to assist; contrast with PFAM which actually utilizes HMMs for the majority of work and is verified afterward by humans.
Certain folds like the beta-Trefoil and TIM Barrel benefit from containing only proteins that cleanly fit into some subcategory or several subcategories– it is then possible to just drill into the right level of categorization and pull out all of the beta-Trefoils and TIM Barrels we want.
The 4-Alpha-Helix Bundle constitutes a fold of protein that manages to be spread around the databases, being a very common secondary structural repeat; also a very small repeat when compared to the two giants above. These two items represent an interesting contrast too. Both machine and human intelligence pulls TIM Barrels together while sprinkling alpha-helix bundles across databases and subcategories. And yes, the size difference helps too.
So, I’m starting with a structural then sequence based alignment for single domain TIM Barrels and alpha-helix bundles; to be completely focused, an objective is named: To identify where sequence repeats occur in each individual protein.
